
The IPA of major gene expression alterations, cooperative game concept ana lysis, chromosomal alterations and microRNA impacted genes exposed the axonal advice since the most significant path way while in the pathogenesis of unfavorable NB. Important expression improvements amongst VHL/SDH and MEN2/NF1 PCC The comparison of VHL/SDH and MEN2/NF1 asso ciated PCC was doable on 3 platforms. To generate a far more reliable dataset of significant expression alterations, we supplemented these gene lists by using a even further one particular, located by literature search. By this technique, we have recognized 240 prevalent considerable gene expression adjustments in at least two gene lists, and among these 9 had been popular in all four comparisons, such as, phenylethanolamine N methyltransferase, ret proto oncogene, Src homology 2 do key containing transforming protein one.
By IPA, IGF1 signaling turned out to get by far the most substantially influenced pathogenic path way concerning SDH/VHL and MEN2/NF1 PCC. Sizeable expression modifications between MEN2A and VHL PCC The comparison small molecule Aurora Kinases inhibitor of MEN2A and VHL associated PCC was feasible on three platforms, as well as a even further dataset was analyzed by SAM, also three substantial gene lists had been col lected from literature search. Through the comparison of those seven gene lists, we’ve got identified 162 genes which were widespread in no less than two gene lists, as an example, CHGB, neural cell adhesion molecule 1, pla cental development factor, PNMT, vascular endothelial development factor. Through the IPA of important gene expression alterations, cooperative game theory examination, chromosomal aberrations and micro RNA impacted genes, quite possibly the most sizeable pathogenic pathway was VEGF and hypoxia inducible factor one alpha signaling.
Cooperative game theory examination The amount of index genes revealed tgf beta 1 inhibitor from cooperative game theory examination was similar to the number of sizeable gene expression changes and showed excellent overlap with them. In average, more than 40% of signifi cant gene expression improvements had been identified as index genes by cooperative game concept examination in different comparisons. The pathway evaluation of index genes al means uncovered the exact same canonical pathways as substantial gene lists and supplemented these with several new members. These observations support the applicability of cooperative game concept within the in silico analysis of gene expression.
Discussion Comparison of NB and PCC with other tumors From the cluster analysis of various tumors  and standard tissues, we’ve got observed that NB and PCC tissues are extra similar to one another than to every other tumor en tity. This observation will not be surprising, as the two NB and PCC are neural crest derived tumors. Through the inspection of gene expression adjustments involving NB, PCC and normal tissues along with other tumors, quite possibly the most characteristic gene expression characteristic of NB PCC is clearly associated towards the noradrenalin biosyn thesis and neural crest cell improvement.
and standard tissues, we’ve got observed that NB and PCC tissues are extra similar to one another than to every other tumor en tity. This observation will not be surprising, as the two NB and PCC are neural crest derived tumors. Through the inspection of gene expression adjustments involving NB, PCC and normal tissues along with other tumors, quite possibly the most characteristic gene expression characteristic of NB PCC is clearly associated towards the noradrenalin biosyn thesis and neural crest cell improvement.
The IPA of major gene expression adjustments, cooperative game th
Reply
 modifications linked with person reactions. For all remedy situations, the 2 most influential gene expression alterations were individuals associated with uptake and phosphorylation of glucose 2.
modifications linked with person reactions. For all remedy situations, the 2 most influential gene expression alterations were individuals associated with uptake and phosphorylation of glucose 2.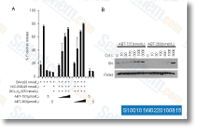 evaluation, Foc TR4 and Foc1 cultures were made use of for inoculating banana roots as de scribed above. At 3 hrs, 27 hours and 51 hrs submit inoculation, the roots of 5 to six banana plantlets subjected on the very same treatment method were pooled together and frozen imme diately in liquid nitrogen for RNA extraction. Serious time quantitative PCR for determination of transcript ranges Complete RNA was extracted from Foc1 inoculated and mock inoculated roots as described over. Initial strand cDNA synthesis was carried out with one.
evaluation, Foc TR4 and Foc1 cultures were made use of for inoculating banana roots as de scribed above. At 3 hrs, 27 hours and 51 hrs submit inoculation, the roots of 5 to six banana plantlets subjected on the very same treatment method were pooled together and frozen imme diately in liquid nitrogen for RNA extraction. Serious time quantitative PCR for determination of transcript ranges Complete RNA was extracted from Foc1 inoculated and mock inoculated roots as described over. Initial strand cDNA synthesis was carried out with one. PLA2 1 accounted for 26. 7% of all transcripts, when PLA2 2 amounted to an extra 5. 5%, The Ovophis transcriptome contained two PLA2 transcripts. even so, the more abundant transcript, PLA2 one, comprised only 0. 65% with the transcriptome, Peptides sequenced by mass spectrometry covered 98. 3% of PLA2 one, but no peptides were uncovered for your small transcript. Serine proteases From the 18 SP transcripts in the Protobothrops library, only two can be confirmed as total, Many transcripts seem to encode dysfunctional SPs. For example, SP16 encodes 36 residues and is bracketed on both ends by halt codons, Offered that it was expressed at a really very low level and that no peptides had been sequenced by mass spectrometry, we feel it’s unlikely to perform any role in envenomation.
PLA2 1 accounted for 26. 7% of all transcripts, when PLA2 2 amounted to an extra 5. 5%, The Ovophis transcriptome contained two PLA2 transcripts. even so, the more abundant transcript, PLA2 one, comprised only 0. 65% with the transcriptome, Peptides sequenced by mass spectrometry covered 98. 3% of PLA2 one, but no peptides were uncovered for your small transcript. Serine proteases From the 18 SP transcripts in the Protobothrops library, only two can be confirmed as total, Many transcripts seem to encode dysfunctional SPs. For example, SP16 encodes 36 residues and is bracketed on both ends by halt codons, Offered that it was expressed at a really very low level and that no peptides had been sequenced by mass spectrometry, we feel it’s unlikely to perform any role in envenomation.